Med tech in a globalized world: overcoming regulatory compliance challenges for international success

Table of contents
- Medical devices marketed in the US
- Some key elements of changes to Part 820:
- EU and medical devices
- Benefits of harmonization and ISO standards
- Global harmonization is driving efficiency
- Harmonization is high on the WHO agenda
- Latin America’s drive towards harmonization
- Harmonization in Africa – Improving healthcare access
- Asia, Pacific and the Middle East / North Africa (MENA)
- Some key general challenges for medical device manufacturers
- Some key challenges for medical device labeling
- Medical Device Regulatory Compliance with TVT

Despite stakeholders’ efforts to harmonize the medical device industry across regulatory jurisdictions, manufacturers face significant challenges in achieving global regulatory compliance. Gradually, however, we are seeing greater recognition globally of quality standards. In regions beyond Europe and North America, harmonization initiatives are well underway.
When the International Medical Device Regulators Forum (IMDRF) gets together twice each year to work on harmonizing regulations and standards, it has 10 direct participants and a handful of forum observers, including the World Health Organization (WHO). But it would be wrong to view representation as small. Another two dozen regulators are affiliate members and numerous regional harmonization bodies and groupings (e.g. the European Union) boost the number of countries involved into three digits.
The sheer number of forums and initiatives worldwide says much about the strong push toward global and regional harmonization. They also tell us much about the urgency, as industry seeks to maximize efficiency, compliance and – especially in poorer regions – access to treatments.
Medical devices marketed in the US
Medical devices are regulated in the US through the Center for Devices and Radiological Health (CDRH). Class III devices are subject to general controls and premarket approval according to 21 CFR 814. Labeling requirements are regulated through 21 CFR Part 801, and quality systems regulation largely through Quality System Regulation (QS regulation) – 21 CFR Part 820. [1]
Recently, a final rule by the FDA aligns the QS regulation with ISO 13485:16, with the aim of promoting harmonization with other regulatory bodies.
Some key elements of changes to Part 820:
- Incorporation of ISO 13485:16. Part 820 has been amended to adopt the quality management system (QMS) requirements outlined in ISO 13485:16.
- Substantial similarity: FDA has determined that ISO 13485:16 provides a similar level of assurance as the previous Quality System (QS) regulation in ensuring device safety, effectiveness, and compliance with the Federal Food, Drug and Cosmetic Act (FD&C).
- Retained scope: The scope of the QS regulation is retained, but many provisions have been amended or clarified for alignment with ISO 13485:16.
- Title change: The regulation is now titled Quality Management System Regulation (QMSR).
The changes do not affect Current Good Manufacturing Practice (CGMP) requirements for combination products, and become effective from February 22, 2026. [2]
This represents a risk-based approach, and is also aligned with ISO 14971:2019 (and amendment A11:2021), which is recognized as a harmonized standard on terminology, principles, and risk management for medical devices.
EU and medical devices
Importantly, the Quality Management System Regulation brings the US closer into alignment with the standards used in the European Union, where medical devices are regulated by the European Union Medical Device Regulation (EU 2017/745) [3] and the In Vitro Diagnostic Regulation (IVDR; EU 2017/746). [4] This has applied since May 2021 for some devices (e.g. new devices), and is being transitioned for many others until 2028. [5]
Medical devices are authorized in the EU by notified bodies in national jurisdictions, not by the European Medicines Agency (EMA) itself, and mutual recognition agreements (MRAs) apply within and beyond the EU (e.g. also in Australia). [6]
A EudraGMDP database serves as the EU-wide repository for manufacturing, import, and wholesale distribution authorizations, as well as Good Manufacturing Practice (GMP) and Good Distribution Practice (GDP) certificates. Since 2011, a public version of the database has been available, providing access to non-confidential information while safeguarding commercially sensitive and personal data. [7]
Other key standards such as ISO 10993-1:2018 (biological evaluation of medical devices) and ISO 14155:2020 (clinical investigation of medical devices for human subjects – good clinical practice) are also driving harmonization worldwide.
Benefits of harmonization and ISO standards
- Streamline product approvals
- Simplify navigation of regulatory landscapes
- Improve global collaboration and market access
- Provide consistent standards for safety, quality, and risk management
- Reduce regulatory overlaps and duplications
- Facilitate trade (e.g. with and within trade groups)
- Enhance product and patient safety
Global harmonization is driving efficiency
When the Global Harmonization Task Force (GHTF) was established in 1992, regulatory bodies and manufacturers saw a glaring need for action. Its founding members were Australia, Canada, the EU, Japan, and the US. In 2012 this disbanded and was replaced by the International Medical Device Regulators Forum (IMDRF), which consists entirely of regulators. As the WHO points out, there are over two million types of medical devices, in 700 device groups. [8] Harmonization doesn’t just help regulators and manufacturers – it offers access to treatments. The following overview is by no means comprehensive, but its gives insight into harmonization moves afoot.
Harmonization is high on the WHO agenda
Impetus for harmonization is coming from many different quarters, and WHO places it high on the agenda. According to WHO, the COVID-19 outbreak increased the momentum, and it led to the creation of a Strategic and Technical Advisory Group on Medical Devices (STAG MEDEV). [9], providing expert guidance.
WHO is also throwing its weight behind best regulatory practices through its Good Regulatory Review document, which establishes a set of principles for medicines and medical devices. [10] Its Good Reliance Practices (GReP) aims at fostering regulatory reliance among different authorities. [11]
Latin America’s drive towards harmonization
Meanwhile, in Latin America, the Colombian, Cuban and Mexican regulatory authorities signed the so-called “Declaration of Acapulco” in April 2023, laying the groundwork for creation of a Latin American and Caribbean Medicines and Medical Devices Regulatory Agency (AMLAC). Again, the COVID pandemic gave initiatives a push here, especially due to disparities in COVID mortality rates in Latin America. A few months later, Argentina, Brazil and Chile lent their support for a common regulatory agency. [12]
Harmonization in Africa – Improving healthcare access
In Africa, the African Medicines Agency (AMA) aims at establishing a coordinated system of approvals across treaty countries. Unfortunately, things got off to a very slow start when the initial groundwork was laid in 2009 by an African Medicines Regulatory Harmonization (AMRH) program. Today, it is gaining momentum. This is in part thanks to substantial funding provided by the European Commission via EMA. Funding has the stated aim of supporting regulatory systems locally, nationally and through an Africa-wide regulatory approach (presumably one similar to EMA’s in Europe). [13]
Asia, Pacific and the Middle East / North Africa (MENA)
Harmonization is being driven in the Asia-Pacific region through an Asia-Pacific Economic Cooperation (APEC) Life Sciences Innovation Forum (networked with the International Medical Device Regulators Forum (IMDRF), and the ASEAN Medical Device directive. [14] In China, although many ISO and International Electrotechnical Commission (IEC) standards have been adopted, these are sometimes based on outdated versions. Labeling requirements also vary in content, with additional information required. (See this dissertation for insights into the Chinese market.) [15]
India was characterized historically by a notable absence of medical device regulation until 2017, ending with the adoption of Medical Devices Rules. [16] (See this excellent overview of regulations in India.) [17]
While many MENA countries adopt international standards and use EMA and FDA regulations as benchmarks, they are marked by divergence. Gulf Cooperation Council (GCC) countries are closely aligned (see this article for more). [18]
Some key general challenges for medical device manufacturers
The global medical device industry operates in an increasingly complex regulatory environment. Manufacturers face several overarching challenges:
- Regulatory discord, and understanding the many different regulations (e.g. labeling requirements)
- Quality and compliance (adopting consistent quality and compliance standards that meet requirements in all jurisdictions)
- Security, cyber-security and data safety (avoiding breaches, observing regulatory frameworks for privacy, especially in increasingly patient-centric environments)
- Regulatory time-lags in regard to technological developments (e.g. regulatory responses to AI in medical devices)
- Workforce challenges (training, workloads)
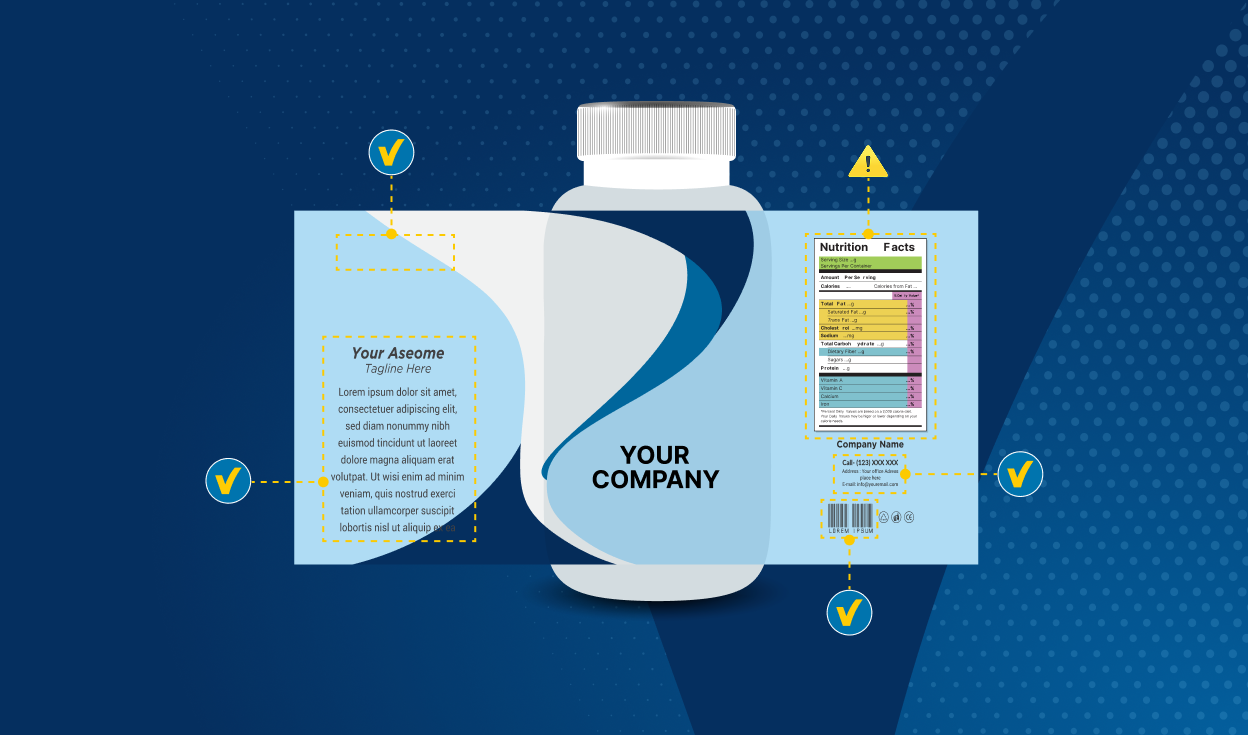
Some key challenges for medical device labeling
Labeling remains a focal point of regulatory compliance, with manufacturers navigating a range of challenges:
- Differing language requirements
This includes multilingual EU versions (as opposed to single-language versions) - E-labeling (many changes are taking place here and harmonization therefore will need to keep pace with the increasing use of e-labeling
- Managing labeling consistency across jurisdictions, while striving for the most universal labeling possible
- Understanding the labeling requirements and keeping up to speed with regulatory changes, more-frequent updates (managing workloads, training, knowledge, skill-sets and staffing, or coordinating across jurisdictions)
- Managing and ensuring the accuracy of dual formats (e-labeling and physical labeling)
- Developing cost-effectiveness measures to reduce expenditure due to local regulations
- Future-proofing labeling workflows to mitigate risk in divergent jurisdictions

A 2024 study commissioned by Schlafender Hase provides further insight into these challenges. The survey of 202 regulatory professionals from Class 2 and 3 medical device companies in Germany and the US underscores just how difficult compliance has become:
- 65% reported that managing translations is a significant hurdle.
- 61% highlighted barcodes as a source of compliance challenges, reflecting the complexities of accuracy and traceability.
- 60% cited graphics, including symbols like device sterilization icons, as particularly tough to manage.
- 59% struggled with tables, emphasizing how even layout elements can complicate labeling workflows.
- 54% found getting the text itself right to be a persistent issue.
These findings reflect the growing need for streamlined, accurate, and adaptable workflows to ensure compliance in a globalized regulatory landscape. Labeling accuracy, consistency, and efficiency have never been more critical to achieving international success.
Medical device regulatory compliance with TVT
Automated proofreading can help you achieve compliance and manage your labeling and documentation workflows more efficiently. Whether you’re navigating pre-market approvals or managing post-market documentation, automated tools, like TVT, offer the precision and efficiency you need to meet stringent medical device regulations. Learn how automated proofreading can help safeguard your compliance efforts while saving time and resources.
Labeling compliance in regulated industries: the complete guide
Labeling compliance is more than checking text. See how regulated companies manage approvals, multilingual content, artwork verification, and regulatory change control.